Design Introduction
This project is structured into two major phases. As a consequence, we use different construct designs for each. The first phase focuses on research aimed at identifying the enzymes that are likely to catalyze the expected reactions, based on a highly refined computational analysis that can be consulted in the DryLab section. This stage involves proposing candidate enzymes and conducting individual laboratory tests for each one. To support this effort, we partnered with Ginkgo Bioworks, a company that sponsored the DNA synthesis of individual plasmids, each encoding one of the enzymes in the pathway. Additionally, they provided experimental testing of these enzymes to validate their activity.
The second phase focuses on the reconstruction of the entire metabolic pathway in yeast. Based on our mathematical model, the pathway is expected to be more efficient if all the necessary enzymes are integrated into a single Saccharomyces cerevisiae strain, rather than distributed across multiple strains with metabolite transfers between them. This consolidated approach allows for better control and optimization of the pathway, reducing inefficiencies. Therefore, our next steps will focus on refining this design for optimal performance.
Saccharomyces cerevisiae is an ideal chassis for this project due to several key factors. It can achieve high cell density in cultures and is a well-characterized eukaryotic organism, which makes it highly amenable to genetic manipulation. Its ability to perform post-translational modifications ensures proper folding and functioning of complex eukaryotic proteins, which is essential for the enzymes involved in our pathway. Additionally, S. cerevisiae's robustness under various environmental conditions, its ease of large-scale cultivation, and its capacity to produce high yields of recombinant products make it a cornerstone of industrial biotechnology. The extensive genetic tools and resources available for S. cerevisiae further enhance its flexibility and efficiency in synthetic biology applications, making it the perfect platform for our pathway integration and optimization.
Phase I: Designs for Testing
In the initial phase of this project, we proposed a set of 11 enzymes that required individual testing to verify their potential to catalyze the desired reactions. To facilitate this, Ginkgo Bioworks offered their support by synthesizing constructs capable of expressing these enzymes in different yeast strains. Since we do not have permission from Ginkgo to share specific details about their constructs, this section does not include them. However, the constructs were transcriptional units consisting of a constitutive promoter, followed by our coding sequences (CDS), and a terminator.
Out of the proposed enzymes, we were particularly interested in testing only those that had not been previously validated in the laboratory, in order to explore their functions. As a result, the CDSs utilized corresponded to enzymes 5, 6, 7, 8, 9, and 10. Each of these CDSs was optimized for yeast codon usage to ensure efficient expression. Our team held several meetings with Ginkgo to assist them in sourcing the required metabolites for each enzymatic reaction.
However, Ginkgo’s experimental assays did not yield conclusive results. Thus, it became our responsibility to perform the enzyme testing ourselves. To proceed, we carried out a secondary enzyme selection process, which can be reviewed in the "Alternative Enzymes Selection" section. This refined selection was further validated through docking analysis and molecular dynamics simulations, as detailed in their respective sections.
Following this refinement, Ginkgo offered to synthesize constructs for each of the alternative enzymes we proposed, resulting in a total of 127 plasmids. These constructs were designed for genomic integration, with codon optimization tailored for yeast. Ginkgo sent these reagents to our university for further testing. It is important to emphasize that the construct designs were entirely the work of Ginkgo, and our contribution was limited to providing the CDS sequences.
Phase II: Designs for Production
Metabolite synthesis by enzymatic reactions is a complex process for all living organisms on Earth. In nature, the synthesis machinery is a system composed of numerous components and variables that influence the final outcome. One of the most important factors to consider in such mechanisms is the time required to complete each reaction.
For example, during a metabolic pathway, if an enzyme is highly expressed but its corresponding substrate is not present in the necessary quantity, the enzyme will not be able to function. As a result, the organism will suffer from inefficient use of amino acids that could have been allocated to more essential tasks. Due to this, evolution has favored the development of various tools that allow cells to optimize when and what proteins are translated to maintain homeostasis. One such tool is reflected in the configuration of transcription networks. A common arrangement in metabolic transcription networks to address this challenge is the creation of temporal programs. These programs allow proteins to be translated approximately at the same time that their corresponding metabolites accumulate. (Alon, 2019).
This is often achieved by the sequential activation of the promoters involved in the pathway, where each promoter responds to the same transcription factor but with varying affinities. This means that some promoters are activated at lower concentrations of the transcription factor, leading to a staggered expression of genes as the activator concentration increases. These configurations are known as Single Input Modules (SIM), where a set of genes is regulated by a single transcription factor. (Alon, 2019).
In synthetic biology, expression systems are often tasked with producing proteins that offer no immediate benefit to the host organism's survival. When multiple genes need to be expressed simultaneously, as in the case of metabolic pathways, the resource-saving mechanisms typically found in natural systems are usually bypassed. Given that our project involves such a pathway, we hypothesize that implementing an artificial temporal program could optimize the production of the necessary proteins. More importantly, simultaneous expression of multiple transgenic proteins could exacerbate pleiotropic effects and increase the risk of autotoxicity from accumulated metabolites. Additionally, issues such as protein aggregation are commonly observed in these scenarios, further underscoring the need for controlled, staggered expression.
Phase II: Designs for Production
A design that mimics nature to optimize productivity
We discovered that three of the enzymes involved in piperamide synthesis are overexpressed in Piper nigrummature fruit. These three proteins participate in the final stages of the pathway, with one of them being Piperamide Synthase, which performs the last step in piperamide synthesis. The fact that evolution has seemingly developed a mechanism to separate the expression of these enzymes over time provided us with a valuable insight into the importance of timing control in this molecular process. Additionally, we were familiar with a known mechanism to achieve this—Single Input Modules (SIMs)—which gave us a solid foundation for our design.
However, implementing a system with 11 promoters activated by the same transcription factor, but with different affinities, presents a significant challenge. Moreover, using 11 transcriptional units would drastically increase the number of DNA parts required, adding complexity to the system and increasing the likelihood of failure.
To address this, we devised a completely artificial temporal program using two promoters: a constitutive promoter to manage the expression of the first block of proteins, and a galactose inducible promoter (pGAL) to activate the production of the final proteins—those that we identified as being overexpressed in the mature fruit—once their substrate demand could be met.
This approach only partially solved the problem, as the number of required promoters, terminators, and ribosome binding sites would still be substantial. While using bacteria as the expression system could have addressed this through polycistronic constructs, bacteria are not entirely reliable for expressing eukaryotic proteins. Given that we were working with 11 proteins, the probability of encountering expression difficulties was high. Therefore, we opted for yeast as our expression system.
At this point, we leveraged a powerful tool: 2A peptides. These peptide sequences can be inserted between two genes, linking them and enabling co-expression through a single promoter. Essentially, they function like polycistronic units. We decided to include them in our design. (Liu, et al., 2017).
The PTE2A construct has been shown to be highly efficient for quad-cistronic constructs in mice, as it successfully expressed a good amount of proteins. However, it did exhibit some challenges in translating the last fragments of the transcriptional unit. (Liu, et al., 2017). Interestingly, we propose that this imperfection could be useful for fine- tuning the timing of the temporal program.
We established the order of protein expression in a manner that reflects the synthesis steps of piperamide, where each enzyme's position is determined by the step it performs in the pathway. The pathway involves two sub- pathways, each starting with an amino acid (phenylalanine and lysine) and converging at Piperamide Synthase.
By combining a constitutive and an inducible promoter, along with adjustments based on the positioning of genes in polycistronic constructs, we have developed a robust mechanism to control the timing of protein production. We organized the genes into three constructs, two of which are managed by a constitutive promoter and one by an inducible promoter. The gene positions were chosen carefully, considering the decrease in expression levels as the distance from the promoter increases. That is also why they were placed into three different transcriptional units, each containing four genes, as including more genes per unit could lead to suboptimal expression.
In the following image we represent above the pathway and below the final design of the artificial polycistronic constructs. It involves two sub-pathways: the phenylalanine sub-pathway, represented by green genes, and the lysine sub-pathway, represented by red genes. These two pathways converge when Piperamide Synthase catalyzes the final reaction to form piperamide. Each gene in the construct is separated by a 2A peptide, which allows the co-expression of multiple genes during translation. Although not depicted in the figure, these peptides ensure that the genes are expressed as individual proteins. The decision to use both a constitutive and an inducible promoter was made to allow for precise control over the timing of gene expression. The inducible promoter can be activated at any chosen time, enabling the creation of a temporal expression program for optimal production.
We revised the design by reducing the constructs from three to two, each containing the coding sequences (CDS) for four genes. The genes are separated by 2A peptides, which allow co-expression of multiple proteins during translation.
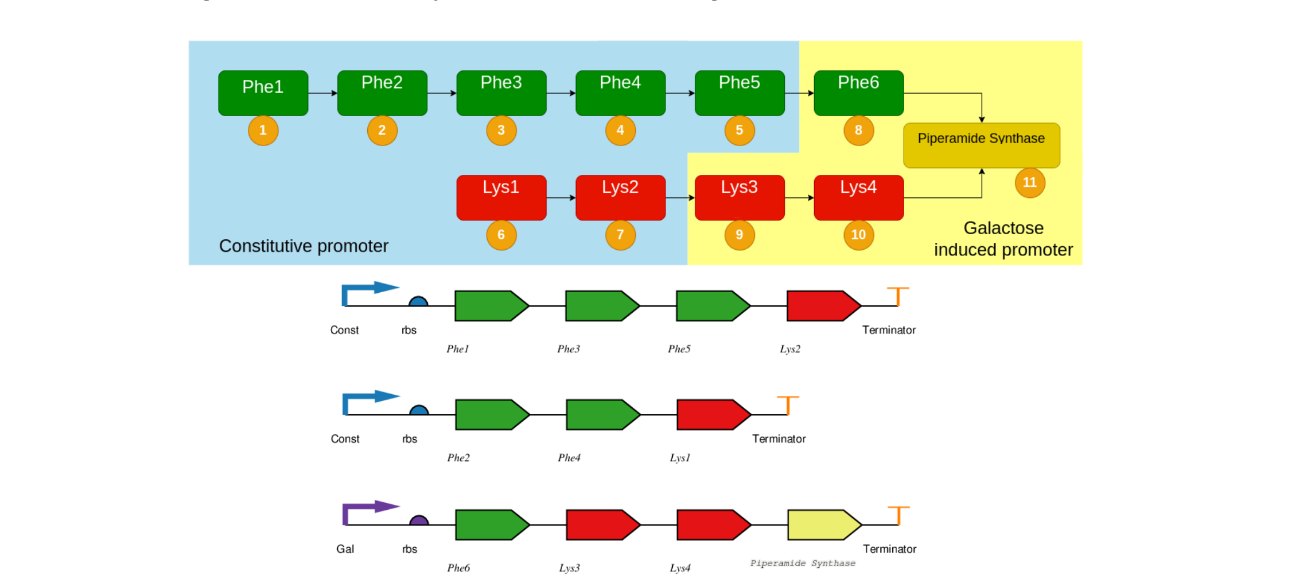
Figure 2: The proposed temporal program is shown at the top. Each number in a yellow circle represents the order the gene is going to be expressed. To accomplish that, we combined the use of a constitutive promoter for the proteins in the blue block and a galactose induced promoter in the yellow block. This last block contains the three proteins that we found out are overexpressed in mature black pepper fruit, except for Lys4, which was not found in the same amount. To polish even more the timing inside each block, we took advantage of the production differences that the 2A peptides imply in artificial poli- cistronic constructions (read the Model section). The bottom shows the design of the artificial polycistronic constructs. Green genes carry out the phenylalanine subpathway, whereas red ones are implied in the lysine subpathway. Both of them come together when piperamide synthase catalyzes the final reaction to form piperamide. Between each gene exists a 2A Peptide that separates two genes during translation (not in the figure). The decision of using a constitutive and an inducible promoter was made because the inducible one can be expressed at any desired time, allowing to create a temporal program.
Problems with our design
The first issue we encountered was discovering that Saccharomyces cerevisiae could naturally produce p-coumaric acid using its own endogenous enzymes (Rodríguez et al., 2015). Since p-coumaric acid is a key intermediate in our pathway, we chose to streamline the design by removing the first two enzymes (1 and 2) to avoid redundancy. Furthermore, a recently published paper revealed that reaction 9 in our pathway was, in fact, a spontaneous process and did not require any enzymatic action (Lv, et al., 2024). As a result, we also removed the enzyme originally assigned to this step. This left us with a total of 8 enzymes, a significant reduction from the 11 initially proposed.
Additionally, the original construct was designed using classical cloning methods with Type II restriction enzymes—a technique that has become quite outdated by modern synthetic biology standards. To streamline the process and align with current practices, we decided to transition to a methodology based on Golden Gate Assembly.
To further enhance the stability of our constructs, we opted for genome-integrating plasmids instead of autonomous plasmids. This approach not only improves the stability of gene expression but is also crucial for biosafety, as it reduces the risk of horizontal gene transfer from these engineered strains compared to using free plasmids.
Revised and optimized designs
Following the investigation and optimization process of our metabolic pathway, we have proposed a new design that consolidates the genetic constructs from three to two. Each construct will contain the coding sequences (CDS) for four genes, with their expression controlled by a modified pGAL1 promoter and flanked by three 2A peptides between the CDS of the proteins, as illustrated in the figure below.

Figure 2: The diagram depicts the composition of the two proposed genetic constructs. Construct 1 contains the CDS for the enzymes of the acyl donor pathway, while Construct 2 includes those of the amine donor pathway. Both constructs share the same promoter, 2A peptides, and terminator, but the genomic insertion site (not shown) will differ.
The GAL1 promoter (Figure 3) was modified based on the BBa_J63006 part, which is similar to the wild-type promoter, by adding 5' and 3' ends compatible with Golden Gate assembly, as used in the Open Yeast Collection. Additionally, the binding sites for the MIG1 protein, which acts as a repressor in the presence of glucose, were deleted (Figure 4). However, the four binding sites for GAL4, the inducer in the presence of galactose, were retained.
We hypothesize that this will allow us to activate the expression of the two constructs by adding galactose to the medium during the stationary phase of growth, while continuing to use glucose as the primary carbon source. This approach will ensure that the concentrations of galactose are not impacted, nor will the yeast's metabolic capacity be compromised by the synthesis of proteins for galactose metabolism.
The endogenous GAL1 promoter will retain the MIG1 binding regions, and thus remain inhibited in the presence of glucose, which is beneficial for controlling the expression of our genetic constructs with a relatively low-cost inducer like galactose.
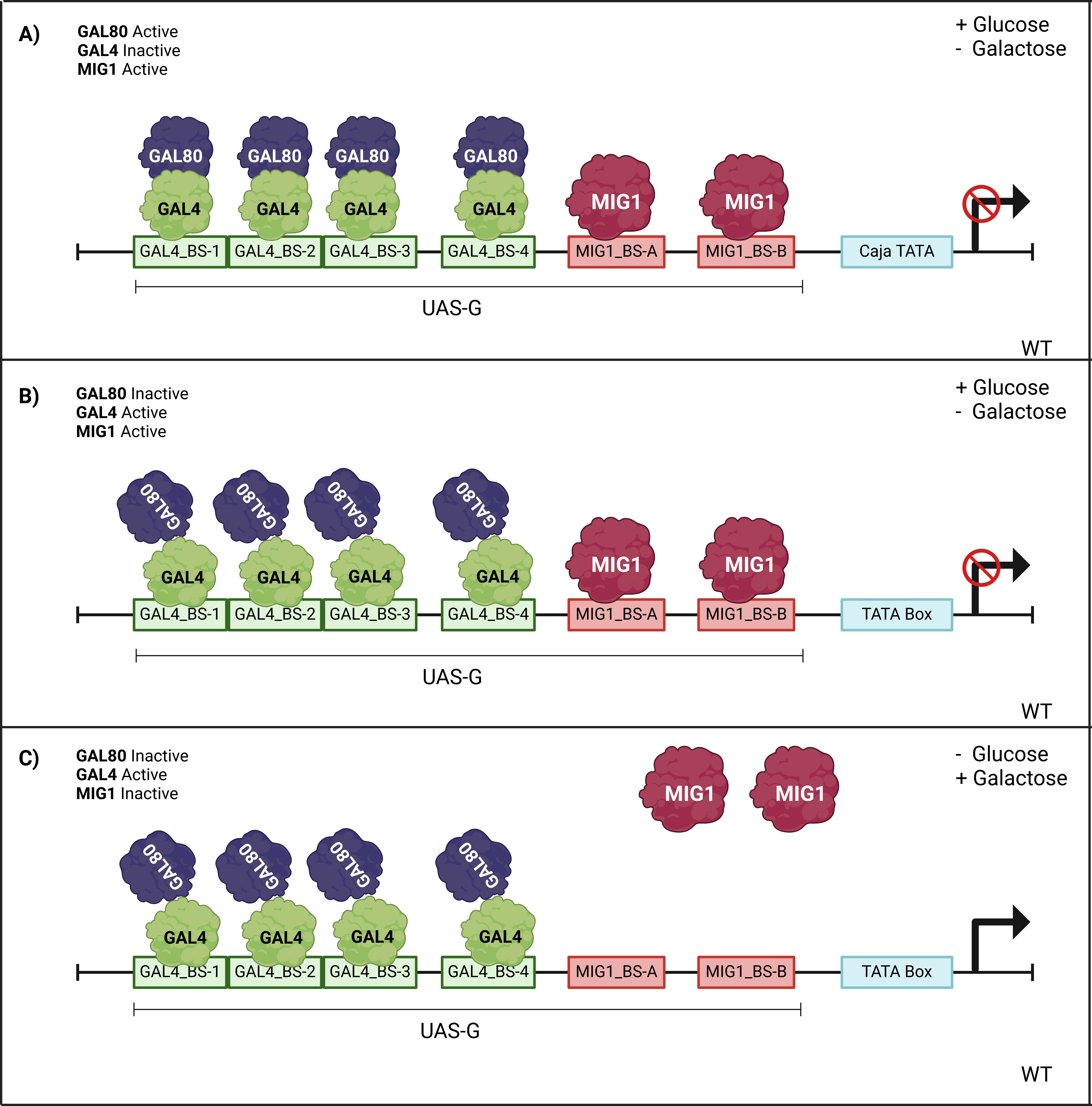
Figure 3: Activation and repression in the UASG region of the wild-type GAL1 yeast promoter under different conditions: a) Glucose present, galactose absent (repressed); b) Glucose and galactose both present (repressed); c) Glucose absent, galactose present (active).
In addition to the deletions of the MIG1 binding sites, the promoter sequences were reviewed to eliminate any restriction sites incompatible with the RFC10 standard. Complementary ends were also added to the promoters to ensure compatibility with other parts of the Open Yeast Collection. We synthesized the promoter sequences accordingly (registered as part BBa_K5479011). To evaluate its performance, we synthesized the GFP enzyme CDS with complementary ends for Golden Gate assembly with other parts from the Open Yeast Collection (Fig 5).
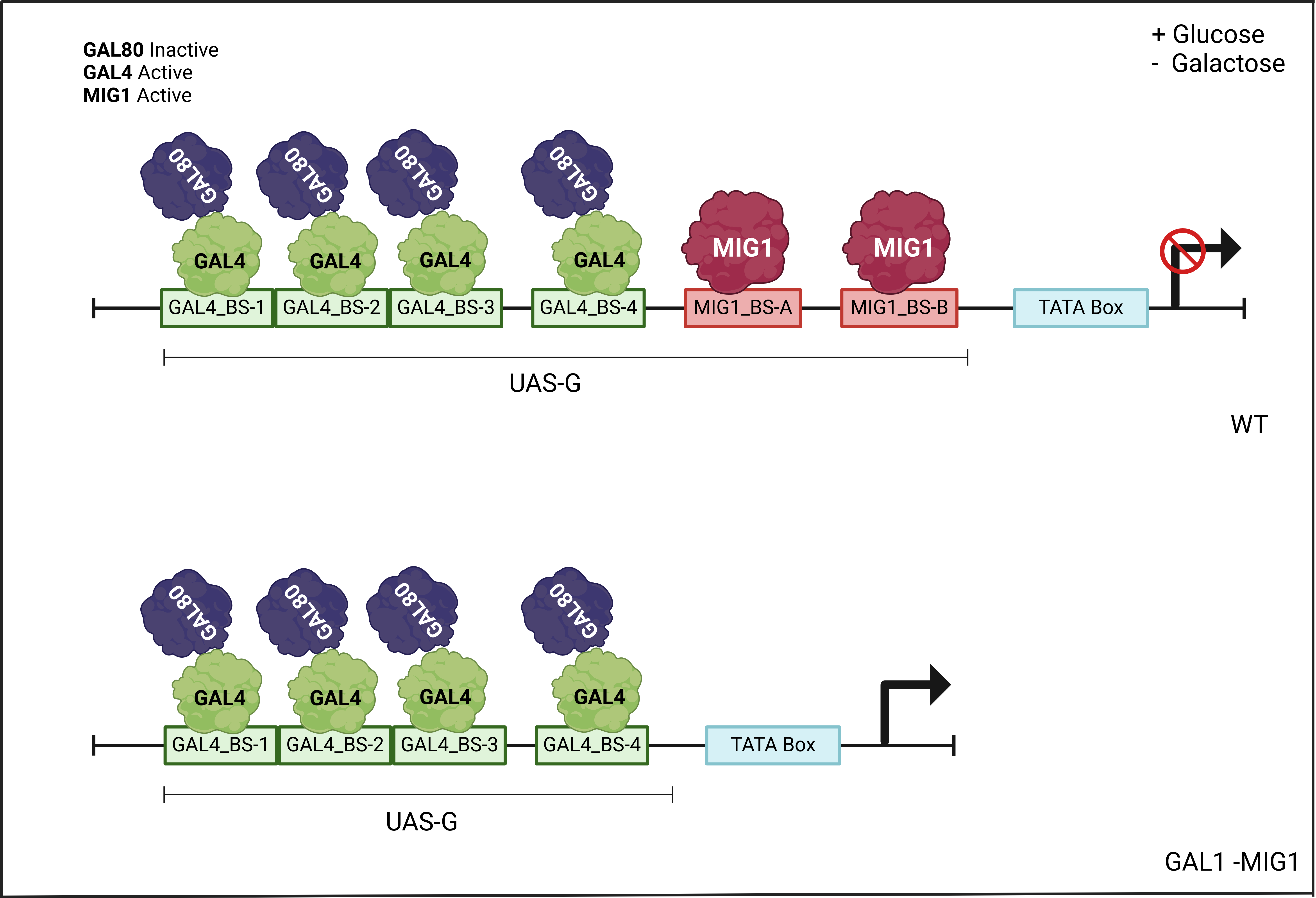
Figure 4: Comparison of the expected behavior of the wild-type and modified (-MIG1) promoter in the presence of glucose and galactose.
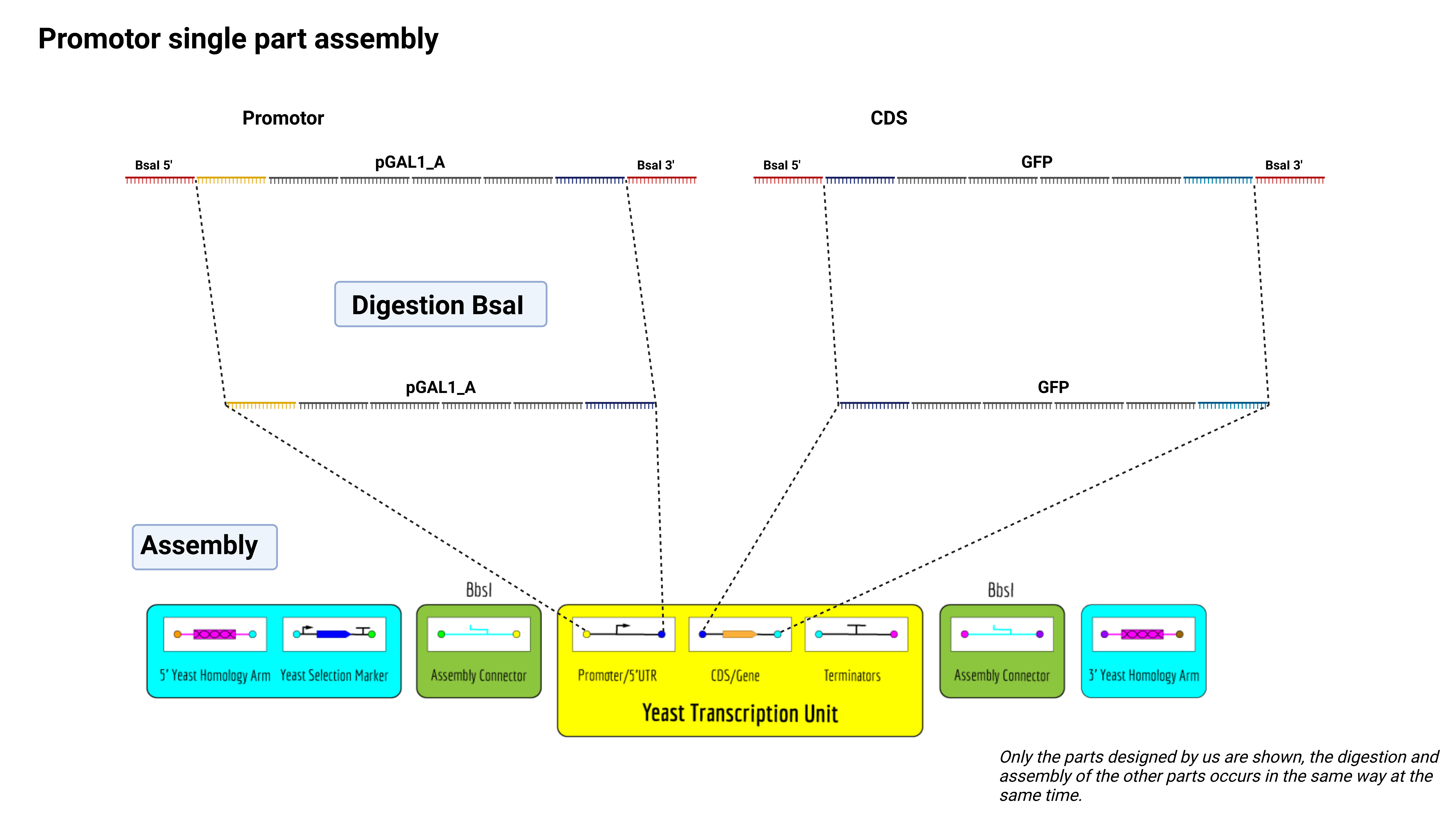
Figure 5: Schematic of the proposed assembly of the modified pGAL1 promoter together with GFP, using Golden Gate.
Alongside the promoter, we also designed and synthesized a part containing the 2A peptide sequences. These sequences were optimized with complementary ends for Golden Gate assembly, ensuring compatibility with other parts used in the Open Yeast Collection. The purpose of this element is to facilitate the use of 2A peptides in assembling our final construct (Fig 6). The selected CDS from the pool synthesized by Ginkgo Bioworks will be amplified using primers that add these complementary ends.
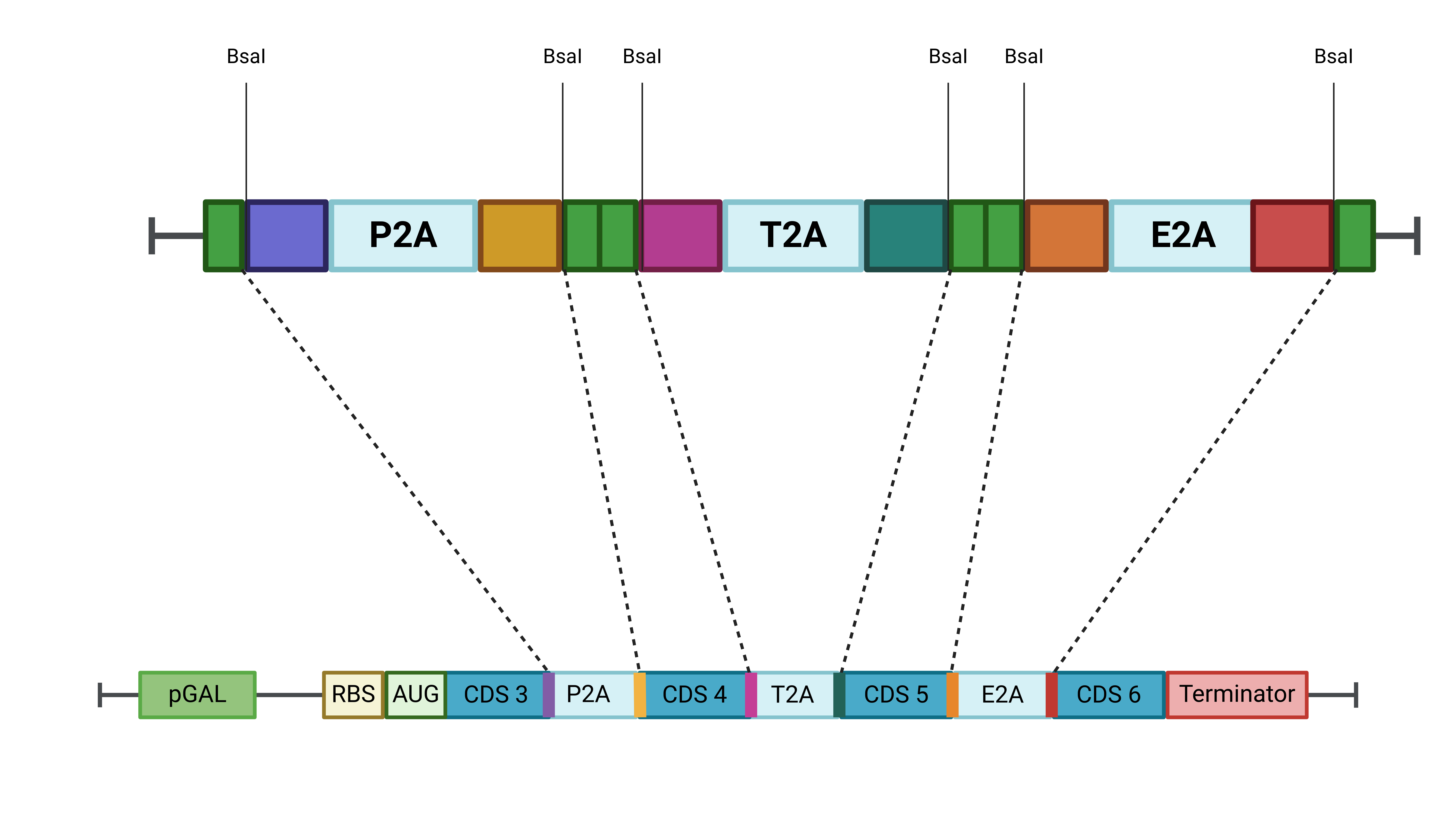
Figure 6: Schematic representation of the designed and synthesized part containing the PSA, T2A, and E2A peptides with complementary ends for Golden Gate assembly.