Differential Expression Analysis
Differential Expression Analysis
In order to find the other enzymes present in the biosynthetic pathway, a differential expression analysis was performed in the same way piperine synthase and other enzymes have been discovered. RNAseq data from four different tissues of P. nigrum were used; these tissues were leaf, flowering spadix, and black pepper fruits at two different stages: 20-30 days post anthesis and 40-60 days post anthesis; each tissue was represented by three biological replicates, having a total of 12 samples. From now on, the spadix of the panicle will be referred to just as panicle. The mature fruit (40 days post anthesis) has the highest piperine and piperamide concentration from all the four tissues studied; considering this, it is expected that the genes over-expressed in mature fruit in comparison with the other tissues are related to piperine/piperidine biosynthesis.
The RNAseq data were obtained from the SRA database with BioProject ID: PRJEB38192; the sequencing technology used was Illumina HiSeq 2000 paired end sequencing. The average number of reads per run was 35.46 Mbp, the quality of the reads was evaluated by using FastQC, a quality control tool for high-throughput sequence data, and summarized with MultiQC. The reads were checked for adapter content and these were removed with Trimmomatic. The sequences were also quality trimmed using a sliding window of four nucleotides and a required average quality of 20, with a minimum length of 125 bp. After the raw data of the samples were cleaned, the average number of reads per run was 27.08 Mbp. The cleaned reads were mapped to the CDS sequences of the reference genome of P. nigrum by using Kallisto.
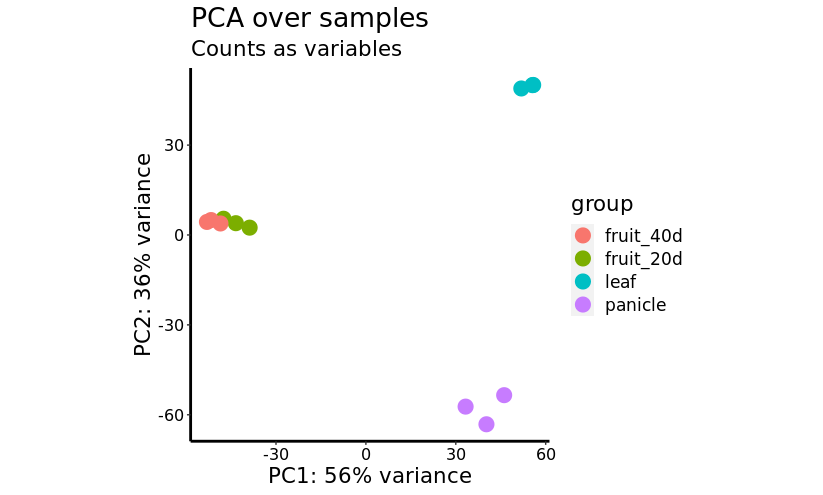
With the count values obtained from the mapping, a differential expression analysis (DEA) was performed using the DESeq2 algorithm, a method for differential analysis of count data, using shrinkage estimation for dispersions and fold changes to improve stability and interpretability of estimates. With this data, a principal component analysis (PCA) was performed, and can be seen in the next figure. The principal component number one, which contains 56% of the variance of the data, can differentiate between both the immature and mature fruit samples and the leaf and spadix tissues; the second component, with 36% of the variance, differentiates clearly between leaf and panicle tissues. Even though there is a distinction between both fruit stages, they cannot be differentiated completely with this analysis, which indicates that there is a lot of similarity in the gene expression of both stages.
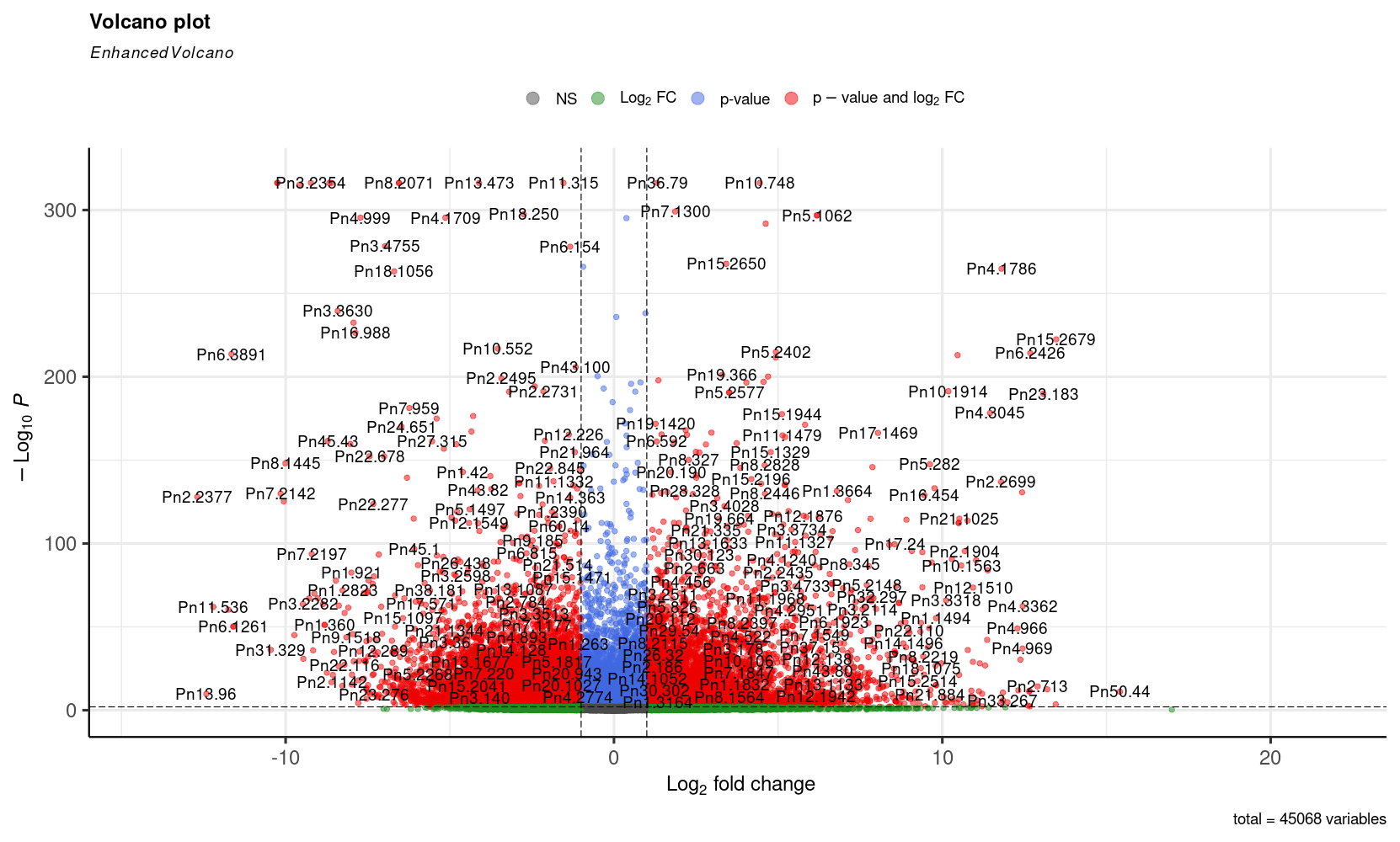
The differential expression analysis found 17,071 differentially expressed genes across the selected tissues; these genes form an expression pattern for every tissue in the analysis. These genes were selected using a likelihood ratio test (LRT) to verify if they were significantly expressed in a different way across tissues. Genes with an absolute log2FoldChange value greater than one and an adjusted p-value lower than 0.05 in comparisons between mature fruit and every other tissue were considered as differentially expressed in mature fruit. These results can be seen in the next figures.
Figures:
Volcano plot of differentially expressed genes in mature fruit
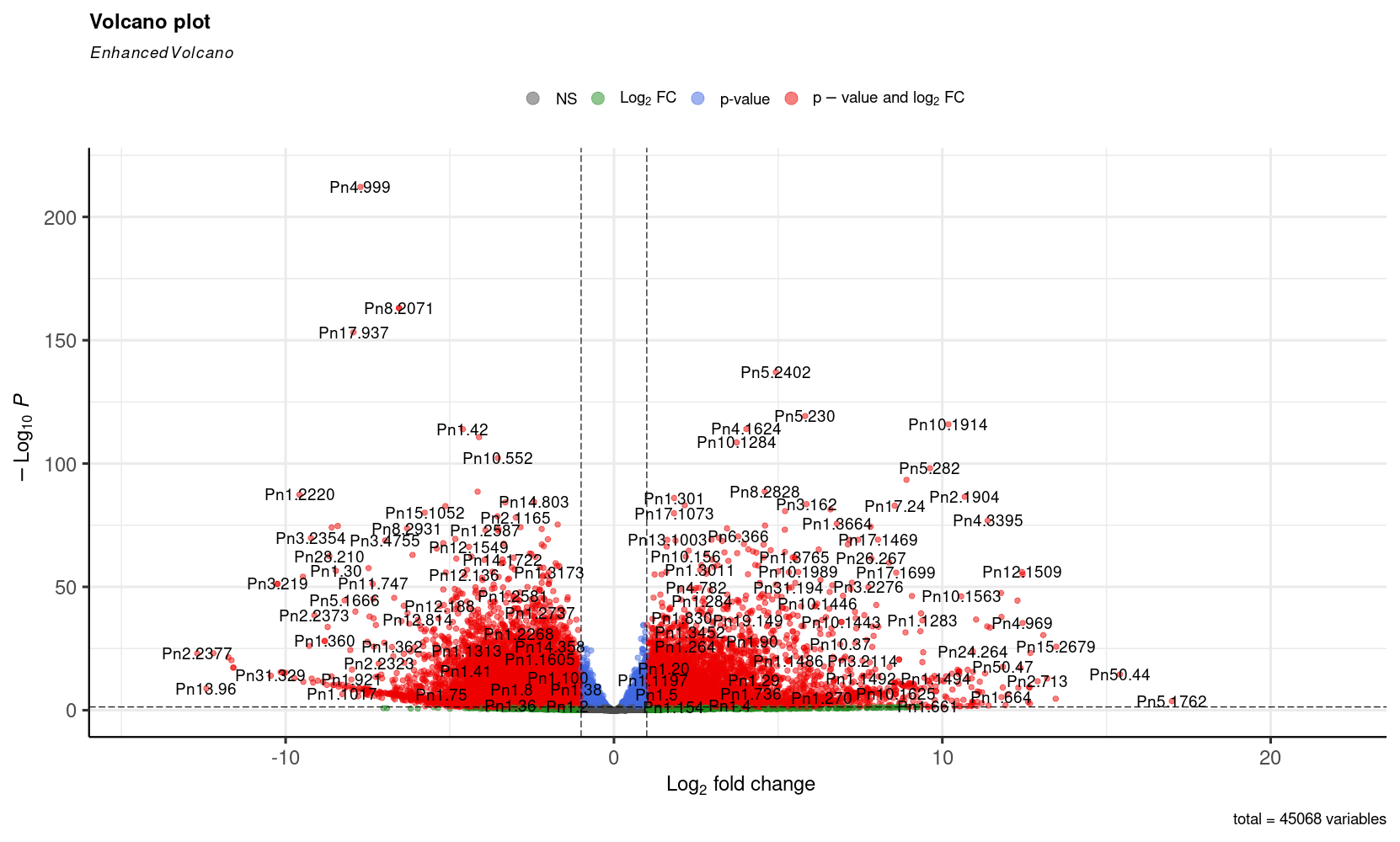
Volcano plot of differentially expressed genes in mature fruit, likelihood ratio test.
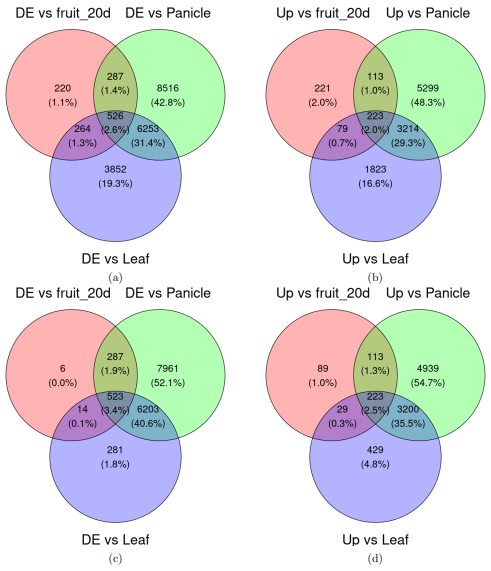
A search for the genes differentially expressed in mature fruit in comparison with every other tissue studied was performed; these results were verified by the LRT with an adjusted p-value lower than 0.01 and an absolute log2FoldChange greater than 1. The results showed that 19,918 genes were differentially expressed in mature fruit in comparison with some tissue, but only 526 were in comparison to all tissues. From these genes, 10,972 were over-expressed in comparison with some tissue and only 223 genes were over-expressed in comparison to all tissues. From these genes, only those selected by hypothesis testing by LRT were conserved; 15,275 genes were differentially expressed in mature fruit in comparison with some tissue, but only 523 were in comparison to all tissues. This represents a reduction of three genes that were not considered as differentially expressed to all tissues after the hypothesis testing. From these genes, 9,022 were over-expressed in comparison with some tissue and 223 genes were over-expressed in comparison to all tissues; here we can see that there was no reduction in the number of genes interpreted as over-expressed to all other tissues, meaning a zero false discovery rate in this group.
Venn diagram of differentially expressed genes in mature fruit. DE: Differentially expressed genes, Up: over-expressed genes. a-b pre hypothesis testing by LRT, c-d post hypothesis testing by LRT.
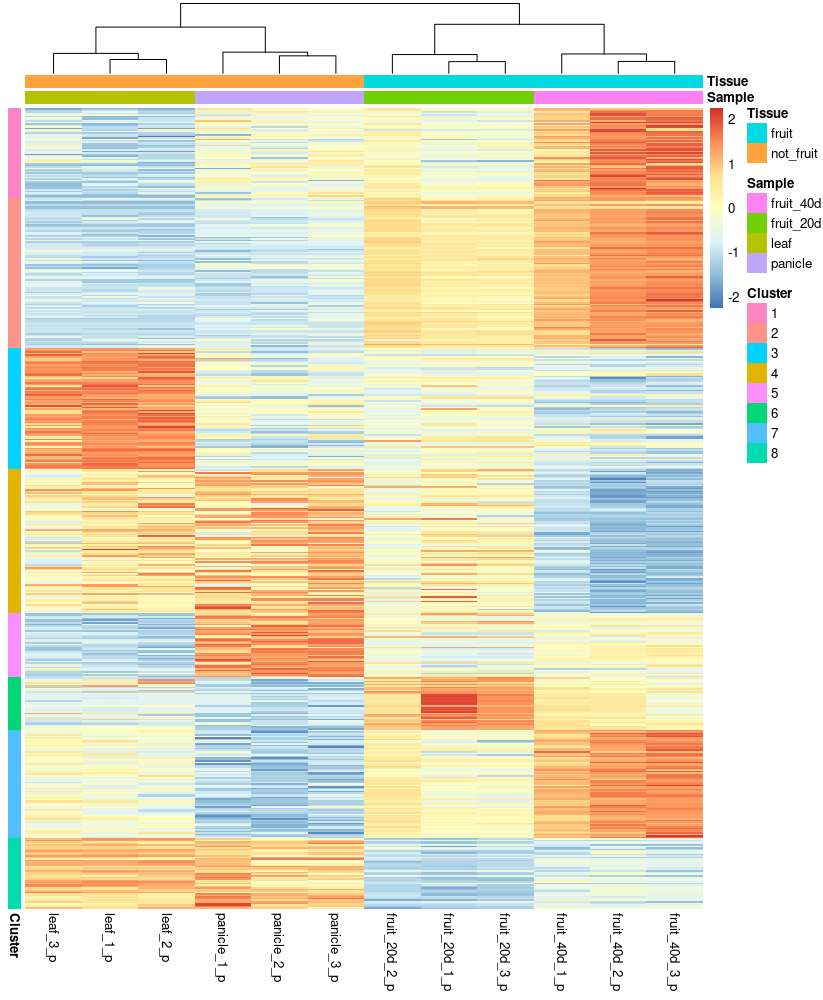
With the 523 genes found that are differentially expressed in mature fruit, a transcriptome profile was made. The heatmap shows the gene expression level for all the samples; the genes are clustered in eight clusters found by k-means, and the number of adequate clusters was selected by the elbow method using the total within sum of squares of the cluster. The clusters 1, 2, and 7 are those of interest since they contain the genes over-expressed in mature fruit in comparison to all tissues, probably involved in the biosynthesis of piperamides.
The second gene most over-expressed in mature fruit is annotated as a terpenoid cyclases/protein prenyltransferases superfamily protein by the GeneFamilyClassifier tool. This gene has the highest log2FoldChange found (12.69) and an adjusted p-value of 9.74 × 10-129. We propose that this enzyme could perform the step nine of the proposed biosynthetic pathway. In order to support this hypothesis, a molecular docking methodology was performed, as with the other proposed enzymes. Even though enzymes 5 and 6 are characterized for the synthesis of piperine, these enzymes cannot act on ligands with two fewer carbons like the proposed piperamide. For enzyme five, the gene Pn7.1626 was selected based on its similarity with the characterized protein (ID: QQS74306), with 76.908% identity and E-value ≊ 0. For enzyme number six, the selected gene was Pn16.1198; the protein produced from this gene is annotated as a 4-coumarate:CoA ligase 2, which is the expected activity for the enzyme. It has a 75.632% identity with the known enzyme (ID: QGY72664), and it is the sixth over-expressed gene in mature fruit with a log2FoldChange of 7.31 and an adjusted p-value of 5.89 × 10-81. Its expression is higher in mature fruit than the known protein, which is the 189th over-expressed gene with a log2FoldChange of 1.84 and an adjusted p-value of 9.78 × 10-7.
The piperine synthase (ID: QUS53100) cannot act on an acyl donor two carbons shorter than its normal substrate, piperoyl-CoA, but a similar enzyme called piperamide synthase (ID: QUS53101) can act on a wider variety of acyl donors, including the 3,4-methylenedioxy cinnamoyl CoA, which is the compound involved in our proposed biosynthetic pathway. The piperine synthase corresponds to the gene Pn12.1813; this gene is annotated as a HXXXD-type acyl-transferase family protein, which is the reaction type performed by the piperine synthase. This gene is over-expressed in the mature fruit in position 95, with a log2FoldChange of 4.77 and an adjusted p-value of 1.19 × 10-20.
The piperamide synthase corresponds to the gene Pn6.2477; this gene is also annotated as a HXXXD-type acyl-transferase family protein. This gene is over-expressed in the mature fruit in position 56, with a log2FoldChange of 5.44 and an adjusted p-value of 1.89 × 10-34.
The three remaining enzymes were selected from the literature. Three of the genes selected were found over-expressed in the mature fruit, where the piperine and piperamides are present in the highest concentration in the plant. This expression pattern can be thought of as a temporal expression program for the biosynthetic pathway; in this way, the different transcriptional units were designed, and the expression pattern of the construct works in a similar way to the temporal model found in the expression pattern of the pathway in Piper nigrum.